The self-consistent-field cycle¶
In this exercise we will look more closely at the SCF cycle, (Fig. 10) and how to monitor and alter its convergence.
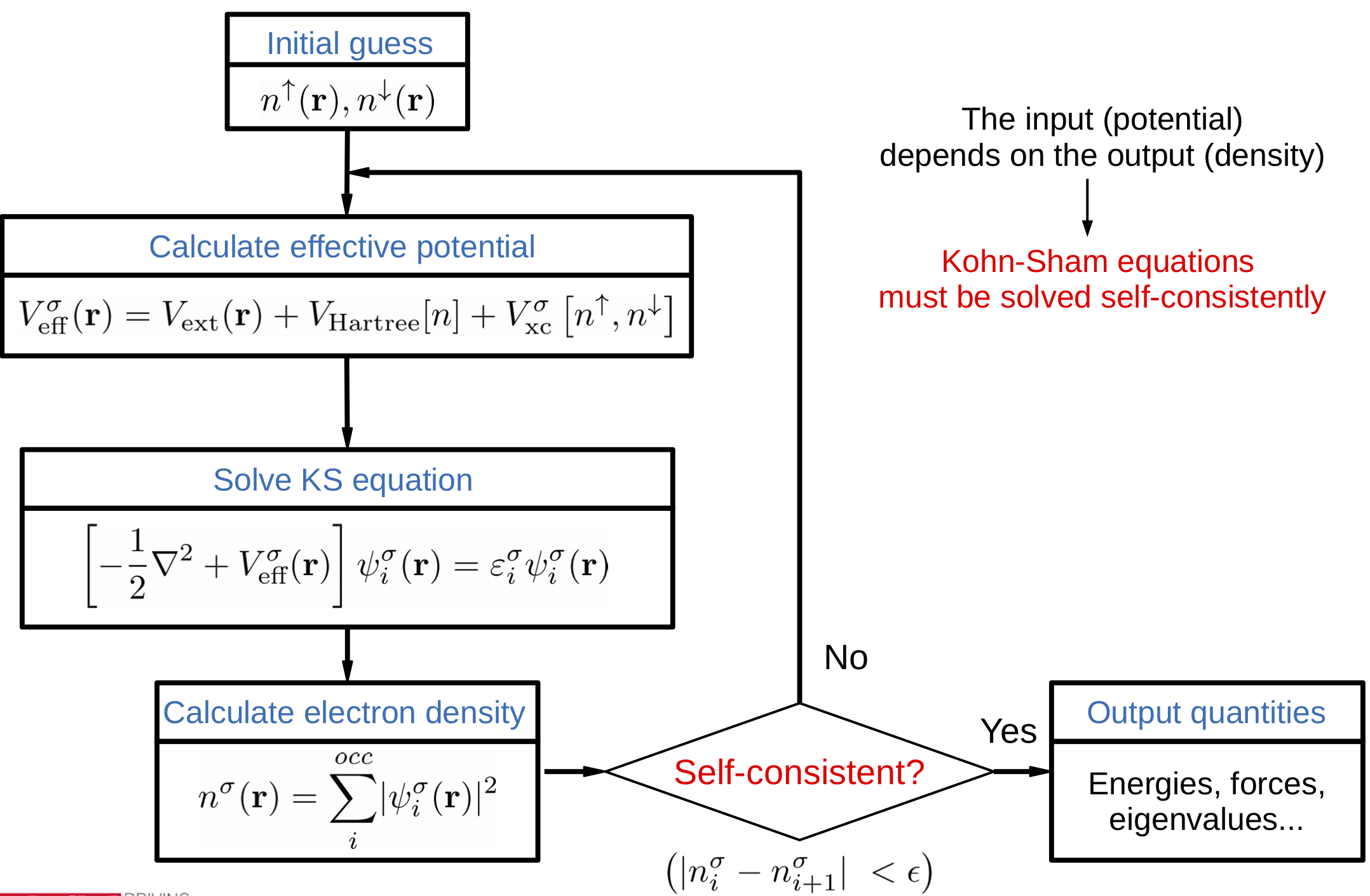
Fig. 10 Flow diagram of the SCF cycle¶
A typical way to accelerate the SCF cycle is to adopt a mixing strategy. This refers essentially to a type of extrapolation, in which we aim for better predictions of the Hamiltonian (or Density Matrix) for the next SCF step.
Whether a calculation reaches self-consistency in a moderate number of steps depends strongly on the mixing strategy used. Choosing the appropriate mixing options for a given system can repay itself handsomely by potentially saving many self-consistency steps in production runs.
In this tutorial we will see a brief summary of the options related to self-consistency, and will practice with them. We strongly suggest to have a look at the manual in case more advanced options are required.
Monitoring the self-consistency¶
There are two main ways in which the SCF condition can be monitored in SIESTA:
By looking at the maximum absolute difference dDmax between the matrix elements of the new (“out”) and old (“in”) density matrices. The tolerance for this change is set by
SCF.DM.Tolerance. The default is 10-4, which is a rather good value, valid for most uses, except when high accuracy is needed in special settings (some phonon calculations, or simulations with spin-orbit interaction).By looking at the maximum absolute difference dHmax between the matrix elements of the Hamiltonian. The actual meaning of dHmax depends on whether DM or H mixing is in effect: if mixing the DM, dHmax refers to the change in H(in) with respect to the previous step; if mixing H, dHmax refers to H(out)-H(in) in the current step. The tolerance for this change is set by
SCF.H.Tolerance. The default is 10-3 eV.
By default, both criteria are enabled and have to be satisfied for the cycle to converge. To turn off any of them, one can use one of the options:
SCF.DM.Converge F
SCF.H.Converge F
Mixing Options¶
SIESTA can mix either the density matrix (DM) or the hamiltonian (H), according to the flag:
SCF.Mix Hamiltonian #{ density | hamiltonian }
The default is to mix the Hamiltonian, which typically provides better results.
Note
Choosing either mixing strategy will slightly alter the self-consistency loop.
With
SCF.Mix Hamiltonian: we first compute the DM from H, obtain a new H from that DM, and then we mix the H appropriately. Then repeat.With
SCF.Mix Density: we first compute the H from DM, obtain a new DM from that H, and then we mix the DM appropriately. Then repeat.
We can then choose the method for the mixing itself, controlled by the
SCF.Mixer.Method variable:
SCF.Mixer.Method Pulay #{ linear | Pulay | Broyden }
with Pulay mixing being the default. The option SCF.Mixer.Weight provides
a damping factor for the mixing when using Pulay and Broyden methods, which is
0.25 by default. In the case of Linear mixing, this means that the new Density
of Hamiltonian matrix (i.e., the one that we are extrapolating) will contain
an 100-X percentage of the previous one (75% for SCF.Mixer.Weight 0.25 ).
The Pulay and Broyden methods are more sophisticated: they keep a
history of previous DMs or Hs (as many as indicated by the
SCF.Mixer.History flag, which defaults to 2).
A simple example¶
In directory CH4 there is a ch4-mix.fdf file very similar to the one in the first-contact tutorial.
Run the example with the provided parameters. You will see that the
program stops with an error regarding lack of SCF convergence: it has
not reached convergence in the allowed 10 SCF iterations (set by the
Max.SCF.Iterations parameter). Before trying anything else you
might want to increase the allowed number of iterations.
Play with the SCF.mixer.weight parameter to see if you can accelerate the
convergence. Also, check the differences when mixing the DM or H.
You have probably noticed that using large values for the weight (close to 1),
reaching convergence becomes extremely difficult or even impossible. However,
if you use a large value, but now set the parameter SCF.mixer.method to
Pulay or Broyden, you will see that the SCF convergence is reached in a few
iterations. Experiment with the values of SCF.Mixer.History and
SCF.Mixer.Weight to see if you can find optimum values
for a fast convergence.
Hint
Always choose the mixing method first, and then worry about
SCF.Mixer.History and SCF.Mixer.Weight.
A harder example (advanced)¶
Directory Fe_cluster contains an example of a non-collinear spin calculation
for a simple linear cluster with three Fe atoms.
The input file fe_cluster.fdf is set up to use linear mixing with a small mixing weight. Check how many iterations are needed for convergence.
Now experiment with other options, and see how much you can reduce the number of iterations.
When you are done, you might want to peruse the file SCFmix.fdf, in which the new mixing technology in SIESTA is exemplified (use of different strategies that can kick in under certain conditions, defined in blocks). You will need to read the manual to follow the meaning of the options.
Note
See that we have commented out the DM.UseSaveDM
option. Otherwise, a new calculation in the same directory would
re-use a (possibly converged, or half converged) DM file.
Note
If you have a hard-to-converge system, you might want to share it with the developers.